Einleitung/ Background
Im Unterschied zu Tieren und Pilzen enthalten pflanzliche Zellen Chloroplasten. Diese Zellorganellen sind die "Biosynthesemaschinen" der Pflanzenzelle. Hier finden die Photosynthese, photosynthetische CO2-Fixierung und weitere wichtige Biosynthese schritte statt. Weniger bekannt - aber für das Verständnis der Funktion dieser Organellen entscheidend - ist die Tatsache, daß Chloroplasten eigenes genetisches Material aufweisen. Diese "extrachromosomale" DNA wird im Unterschied zum (Kern) Genom auch als "Plastom" bezeichnet. Es handelt sich dabei um doppelsträngige, ringförmig geschlossene "plasmidähnliche" Moleküle; pro Organell können zwischen 10 und mehr als 100 dieser (identischen) "cpDNA"- Moleküle vorliegen. Da die Zelle ca. 100 Chloroplasten besitzen kann, ist die Gesamtmenge zwischen 1000 und 10000 cpDNA- Moleküle, d. h. ca. 20% der gesamten zellulären DNA. Die geringe Größe und hohe Kopienzahl macht die cpDNA zu einem günstigen Objekt für molekulare Analysen. Für mehrere Pflanzen liegt bereits die komplette Sequenzinformation dieser Moleküle vor; diese kann aus Sequenzdatenbanken abgerufen werden und für detaillierte Genanalysen verwendet werden. Im diesem Kurs soll eine solche - in der heutigen molekular orientierten Biologie häufige - Analyse am Beispiel eines besonders eingehend untersuchten Chloroplastengens durchgeführt werden.
Das psbA Gen auf der Chloroplasten- DNA codiert das sog. D1- Protein, eines der Reaktionszentrumsproteine von Photosystem II. Dieses physiologisch wichtige Protein hat u. a. auch deshalb große Beachtung gefunden, weil es den intrazellulären Bindungs- und Wirkort für bestimmte Herbicide vom "Diuron"- Typ darstellt (z. B. Atrazin). Das psbA- Gen wurde bereits aus einer Vielzahl von Pflanzen cloniert und sequenziert, darunter auch aus Arabidopsis thaliana, einer kleinen kreuzblütigen Pflanze, die heute als "Modell" in der experimentellen Pflanzenforschung große Bedeutung besitzt.
Für das Verständnis der genetischen Informationsspeicherung und Genexpression ist die Kenntnis der DNA- Sequenz des Genoms eines Lebewesens eine essentielle Voraussetzung. Am Beispiel des plastidären psbA- Gens aus Arabidopsis thaliana soll gezeigt werden, wie eine DNA- Sequenz mit Hilfe moderner molekularbiologischer Methoden ermittelt und anschließend durch computergestützte Analyse auf Promotor und Genstrukturen untersucht werden kann.
Klonierung
Restriktionsendonucleasen (oder Restriktionsenzyme) sind bakterielle Enzyme, die in beiden Strängen eines DNA- Moleküls innerhalb spezifischer Basensequenzen PhosphodiesterBindungen spalten. Viele dieser Erkennungssequenzen sind Palindrome: Auf beiden Strängen von 5'( oberer Strang "vorne") nach 3' (" hinten") ergeben sie die gleiche Sequenz. Durch die Restriktion erhalten die entstehenden DNA-" Fragmente" entweder "überhängende" oder "glatte" Enden. Diese kann man mit Hilfe von des Enzyms DNA- Ligase schließen und somit DNA- Fragmente unterschiedlicher Herkunft miteinander verbinden.
Die Elektrophorese ist ein biochemisches Trennverfahren, bei dem die Wanderung von geladenen Molekülen in einem elektrischen Feld zu deren Trennung ausgenutzt wird. In der hier durchgeführten Elektrophorese verwenden wir eine Gelmatrix aus Agarose. Dies ist ein lineares, langkettiges, unverzweigtes Polymer der Agarose.
Sequenzierung:
Nach Klonierung eines Gens in einen bakteriellen Vektor kann die DNA z. B. nach der meistbenutzten "Sanger"- Methode, die auch als "Kettenabbruch"- oder "Didesoxynukleotid"- Verfahren bezeichnet wird, sequenziert werden. Bei der Sequenzierung werden die entstehenden DNA- Fragmente radioaktiv markiert und anschließend auf einem denaturierendem Polyacrylamidgel ihrer Größe nach aufgetrennt. Nach Autoradiographie ist die Basensequenz direkt auf dem Röntgenfilm ablesbar.
Die DNA- Sequenzierung ist mittlerweile unter Einsatz nicht- radioaktiver Markierungsmethoden automatisiert worden. Hierbei wird jedes Didesoxynukleotid mit einem anderen Fluoressenzfarbstoff markiert, der in einem hochauflösenden Polyacrylamidgel durch Bestrahlung mit einem Argon- Laser photometrisch erfaßt und computergesteuert analysiert.
Materialliste
Laborrrechner (PC), Sequenzanalyseprogramme (z. B. PC_ GENE; IntelliGenetics, USA), Internetadressen von Sequenzdatenbanken (z. B. EBI, NCBI); Gel- Photos, Röntgenfilm- Aufnahmen und Sequenzausdrucke für die Analyse des zu untersuchenden Gens
Durchführung und Auswertung
Ermittlung und Analyse einer DNA- Sequenz am Beispiel des plastidären psbA- Gens aus Arabidopsis thaliana
Sequenz lesen - mit Vektor vergleichen (pBSIKSCx) - A. thaliana Teilsequenz
(psbA) mit vollständiger Sequenz vergleichen - Sequenz ausdrucken
- Sequenzanalyse: - "Restriktionskartierung" aufgrund der Sequenzdaten
(durch "Praxis" = echte Spaltmuster bestätigen) - Restriktionsgele
- Leserahmen bestimmen - AS- Sequenz übersetzen - Promotor suchen
- Ribosomenbindestelle lokalisieren - zuletzt EMBL/ EBI-" Datenblatt" als
Zusammenfassung der Sequenzanalayse ausfüllen.
Allg. Literatur
Westhoff, P., Jeske, H., Jürgens, G., Kloppstech, K., und Link, G. (1996). Molekulare Entwicklungsbiologie -- Vom Gen zur Pflanze (Stuttgart: Thieme Verlag).
Sambrook, J., Fritsch, E. F., und Maniatis, T. (1989). Molecular Cloning:
a Laboratory Manual (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory
Press).
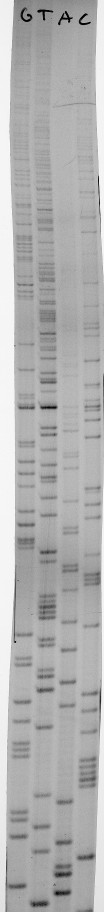
Autoradiographie des verwendeten Sequenzgels: Bereich des psbA-Gens aus Chloroplasten von Arabidopsis thaliana (Original M. Kestermann).
---------------------------------------------------------
G-Block: Pflanzliche Zellphysiologie und Molekularbiologie
Wintersemester 1999/2000
(A. Homann, K. Ogrzewalla, H. Summer und G. Link)
Dieser 2-wöchige G-Block soll zum Verständnis der molekularen und genetischen Grundlagen der modernen Pflanzenforschung beitragen. Das Versuchsprogramm zeigt exemplarische Aspekte der pflanzlichen Molekular-, Zell- und Entwicklungsbiologie mit Bezug zu aktuellen Forschungsthemen auf. Ein besonderes Anliegen dieses G-Blocks ist es, die leistungsfähigen Techniken der heutigen Biologie nahezubringen. Dies soll den Übergang von den Grundblöcken zu der projektorientierten Arbeitsweise der Spezialblöcke erleichtern und damit zu einer Intensivierung des Hauptstudiums beitragen. Die experimentelle Arbeit in kleinen Gruppen wird durch methodenorientierte Seminare und Labor-Demonstrationsversuche ergänzt. Nach Abnahme der Versuchsprotokolle und einem Abschlußkolloqium wird ein Leistungsschein ausgegeben. Begleitende Vorlesung: "Pflanzliche Molekular-, Zell- und Entwicklungsbiologie" (Link).
Die Idee zu diesem Block entstand aus unserer Erfahrung im Hauptstudiengang Biologie und dem Wunsch vieler Student(innen), die Zeit zwischen den experimentellen Grund- und Spezialblöcken sinnvoll und effizient zu überbrücken und optimale Voraussetzungen im Hinblick auf die weitere Ausbildung in den molekularen Pflanzenwissenschaften zu schaffen (z.B. Diplomarbeit!). Dieser im Sommersemester 1997 von uns erstmals angebotene G-Block ist in gewisser Weise ein Experiment, was die Inhalte anbetrifft. Wir haben uns für drei methodisch wichtige Aspekte entschieden, die in der heutigen Pflanzenforschung eine große Rolle spielen: (I) die gentechnische Analyse pflanzlicher DNA, (II) Untersuchungen der Genexpression auf der Ebene der RNA, (III) biochemische Techniken zur Analyse subzellulärer Fraktionen und Proteine. Für niemanden wird (und sollte) der gebotene Stoff völlig neu sein, denn die Analyse von DNA, RNA und Proteinen ist Gegenstand auch anderer G- und S-Blöcke. Uns kommt es auf die Anwendung dieser Techniken im spezifischen Kontext pflanzlicher Organismen an (z.B. Keimlingsentwicklung; Lichtwirkung auf Pflanzen). Auch ist zu bedenken, daß ein und dieselbe Technik in verschiedenen Laboratorien ganz unterschiedlich gehandhabt werden kann. Die größere Souveränität im Umgang mit den sich schnell weiterentwickelnden molekularen Techniken kommt somit der weiteren Ausbildung und ggf. anschließenden wissenschaftlichen Arbeit zugute.
Zur Laborsicherheit: Sie alle haben bereits experimentelle Grund-Blöcke absolviert und sind mit den Grundregeln des sicheren Arbeitens vertraut gemacht worden. Auch in unserem Kurs steht die Arbeitssicherheits-Einweisung vor Beginn des Experimentalprogramms. Hier vorab kurz die DREI GRUNDSÄTZE ZUM SICHEREN ARBEITEN: (1) Nicht unüberlegt experimentieren, sondern nur nach Anleitung und nach gründlichem Lesen und Verstehen des Skripts; ggf. Betreuer fragen! (2) Persönliche und technische Schutzeinrichtungen benutzen: Laborkittel, Schutzbrille, Handschuhe, Pipettierhilfen (NICHT mit dem Mund pipettieren!); Abzug, falls erforderlich (siehe Skript!). (3) Falls dennoch etwas technisch Unvorhergesehenes oder sogar ein Unfall eintritt: Ruhe bewahren und daran mitwirken, daß technische und persönliche Hilfs- und Rettungsmaßnahmen unverzüglich eingeleitet werden können.
Im Skript werden Sie eine Vielzahl von Hinweisen auf mögliche spezifische Gefahrenpunkte der durchzuführenden Experimente finden (z.B. VORSICHT!!! etc.). Diese Hinweise auf MÖGLICHE Gefahren für die menschliche Gesundheit (NUR bei NICHT sachgerechtem Experimentieren!!!) sind nicht zu verwechseln mit Hinweisen auf Gefahren, die nicht den Experimentatoren, sondern den verwendeten Geräten und Materialien bei unsachgemäßer Handhabung drohen. Bitte nehmen sie diese Hinweise genauso ernst, denn es hängt der Erfolg der Versuche davon ab und Geräte und Material sind teuer und werden uns NICHT ersetzt!
Die "Laborsprache" der Molekularbiologen enthält viele Ausdrücke, die nicht unbedingt "lehrbuchreif", aber kurz und prägnant sind. Wir haben versucht, Probleme dadurch zu umgehen, indem wir bei der ersten Erwähnung Anführungszeichen gewählt haben ("Blotten" etc.).
1. Tag
- Allgemeine Einführung und Sicherheitsbelehrung
- Ansetzen und Autoklavieren benötigter Puffer und Lösungen
(Details bei den einzelnen Versuchsteilen).
I DNA-Versuchsteil
II RNA-Versuchsteil
III Protein-Versuchsteil
ab 2. Tag
Spezifische Versuchsvorschriften (I,
II, III):
I DNA-Versuchsteil (A. Homann)
Lösungen am 1. Tag (Montag) ansetzen und, falls angegeben, autoklavieren (Vorsicht! Nur unter Anleitung)
a) 10x SEB (1l):
0,4 M Tris
82 mM NaAc
3,8 mM NaEDTA
-> pH 7,8 einstellen (nur nach Einweisung)
-> autoklavieren (Vorsicht!!!)
b) Extraktionspuffer
CTAB (Hexadecyltrimethyl-
ammoniumbromide) 2 g
1,5 M Tris (pH 8.0) 6,7 ml
250 mM EDTA (pH 8,0) 8 ml
5 M NaCl 28 ml
-> ad 80 ml
-> autoklavieren
Polyvinylpyrrolidon 1 g
Mr 40,000
-> ad 20 ml
-> extra autoklavieren
Beide Lösungen nach dem Autoklavieren vereinen => 100 ml!
c) 8 M LiCl: 20 ml
-> autoklavieren
d) Denaturierungspuffer (1l):
g/l Endkonz.
NaOH 20 0,5 M
NaCl 87,5 1,5 M
e) Neutralisierungspuffer pH 7,0 mit konz. HCl einstellen (1l):
g/l Endkonz.
NaCl 175 3 M
Tris 60,7 0,5 M
f) 2 x SSC
20 x SSC 1:10 verdünnen
g) Eppendorfgefäße und Spitzen -> autoklavieren
h) PCR-Spitzen + Eppis
2. Tag (Dienstag)
Versuch Nr. 1:
Restriktionsanalyse eines unbekannten Plasmids
Das Ziel des Versuches ist es, die Positionen der Schnittstellen mehrerer Restriktionsendonucleasen innerhalb eines Moleküls zu "kartieren".
Restriktionsendonucleasen (oder Restriktionsenzyme) sind bakterielle Enzyme, die in beiden Strängen eines DNA-Moleküls innerhalb spezifischer Basensequenzen Phosphodiester-Bindungen spalten. Viele dieser Erkennungssequenzen sind Palindrome: Auf beiden Strängen von 5' nach 3' ergeben sie die gleiche Sequenz.
Durch die Restriktion erhalten die DNA-Fragmente entweder überhängende oder glatte Enden. Diese kann man mit Hilfe von DNA-Ligase schließen, wodurch man DNA-Fragmente unterschiedlicher Herkunft miteinander verbinden kann.
AUFGABE:
Die Praktikanten teilen sich in 2-4 Gruppen auf. Die Position der folgenden Restriktionsenzyme soll bestimmt werden: EcoRI, PstI, BamHI, XbaI, HindIII, XhoI.
Für die Restriktion soll jeweils ca. 0,5 µg unbekanntes Plasmid (Nr. 324, 0,4 µg/µl), 1X Reaktions-Puffer* und 2 Units (= Aktivitätseinheiten; 1 U = Menge an Enzym, die 1 µg DNA in 1 h schneidet) Restriktionsenzym in einem Gesamtvolumen von 10 µl geschnitten werden:
Beispiel eines Pipettierschemas:
Plasmid 0,4 µg/µl 1 µl
10X Reaktionspuffer 1 µl
Enzymverdünnung 1 µl
H2O ad 10 µl 7 µl
* Der einzusetzende Puffer richtet sich nach dem Enzym bzw. nach den Enzymen: PstI, XbaI, HindIII und XhoI schneiden in Puffer 2, EcoRI und BamHI in Puffer 3.
Es soll mit allen Enzymen jeweils einzeln geschnitten werden, und zusätzlich sollen folgende Doppelrestriktonen durchgeführt werden: EcoRI und BamHI, XbaI und XhoI, XbaI und HindIII, PstI und XhoI, EcoRI und PstI, BamHI und HindIII. Zu beachten ist, daß PstI und HindIII im Puffer 3 nur zu 50 % schneiden. Die Reaktionsansätze sollten auf Eis pipettiert werden, anschließend:
-> 1 h 37 °C **
-> + 2,5 µl 5X Ficoll-Mix pro Probe
-> Ansätze und 5 µl 0,1 µg/µl Lambda-Marker
(23 kb; 9,4 kb; 6,6 kb; 4,4 kb; 2,3 kb; 2 kb + 0,56 kb)
auf das Gel auftragen
1x 1µl unrestr. Plasmid + 2µl 5x Ficoll-Mix + 7 µl H2O
-> Agarosegel-Elektrophorese (1 h 80 Volt)
** Während der Inkubationszeit soll die Gelelektrophorese zur Auftrennung der entstandenen DNA-Fragmente vorbereitet werden.
Die Elektrophorese ist ein biochemisches Trennverfahren, bei dem die Wanderung von geladenen Molekülen in einem elektrischen Feld zu deren Trennung ausgenutzt wird. In der hier durchgeführten Elektrophorese verwenden wir eine Gelmatrix aus Agarose. Dies ist ein lineares, langkettiges, unverzweigtes Polymer der Agarobiose.
Die Agarose wird 0,7%ig in 1X SEB-Puffer angesetzt [z.B. 0,7 g Agarose + 100 ml 1X SEB (1:10 verdünnt aus 10X SEB) in 300 ml Erlenmeyerkolben -> mit Klarsichtfolie abdecken -> 4 min in der Mikrowelle -> unter vorsichtigem Schwenken unter dem Wasserhahn abkühlen -> Gel gießen].
Durch die gewählte Agarose-Konzentration ist eine Auftrennung im Bereich von 0,8 - 10 kb möglich.
Nach dem Lauf wird das Agarosegel in einer Ethidiumbromidlösung
(Konz. 100 µl 10 mg/µl EtBr in 500 ml H2O) gefärbt (ca.
10 min). Durch Bestrahlen des Gels mit UV-Licht fluoresziert die DNA im
sichtbaren Licht (Foto).
Ethidiumbromid ist eine nicht-sequenzspezifisch DNA-bindende Substanz, die sich zwischen einzelne Basenpaare eines doppelsträngigen DNA-Moleküls einlagern kann.
(Achtung: ein Mutagen!).
Zur Auswertung:
Anhand des aufgetragenen Lambda-Markers können die Größen der entstandenen Restriktionsprodukte bestimmt werden.
Die Auftragung des Logarithmus der Fragmentlänge gegen die Laufstrecke ergibt in einem bestimmten Größenbereich eine Gerade!
Ein kleiner Tip: Es handelt sich um einen Klonierungsvektor, der
ein inseriertes Fragment enthält. Damit sind also typische Merkmale
von Klonierungsvektoren, wie eine multiple Klonierungsstelle ebenfalls
vorhanden.
Versuch Nr. 2: 2. (Dienstag) (spätestens 5. Tag: Freitag)
Isolierung von Gesamt-DNA aus Senf-Keimlingen
Diese Methode dient zur schnellen DNA-Isolierung aus kleinen Gewebemengen.
Pro Gruppe soll der Versuch mindestens zweimal (parallel) durchgeführt werden.
- 5 g Kotyledonen (Senfkeimlinge, 4-5 d alt, im Dauerlicht angezogen) in flüssigem N2 mörsern (Achtung: dicke Handschuhe, Schutzbrille, besser: Gesichtsschirm!); NICHT auftauen lassen!
-> Pulver in ein SS34-Zentrifugenröhrchen überführen
-> + 3,5 ml Extraktionspuffer
+ 3,5 ml 8 M LiCl
-> durch vorsichtiges Schwenken mischen und "auflösen"
-> 5 min 65°C, ab und zu schwenken
zweimal wiederholen:(unter dem Abzug durchführen)
-> + 7 ml Chloroform/Isoamylalkohol 24:1 (VORSICHT!)
-> mischen
-> 2-5 sec "vortexen" (rotierend schütteln; Vortex-Mixer)
-> kurz zentrifugieren 3000 rpm (3-5 min)
-> "Überstand" (= obere, wässrige, Phase) vorsichtig in neues
Röhrchen überführen (Achtung: ohne Chloroform!)
zweimal wiederholen: (unter dem Abzug durchführen)
-> (ev. Volumenvergrößerung mit sterilem A. bidest. vornehmen)
-> + 1 Volumen Phenol/Tris gesättigt
-> mischen, zentrifugieren (wie oben)
einmal: (unter dem Abzug)
-> + 1 Volumen Chloroform/Isoamylalkohol
etc. s. o.
=> ca. 5 ml Überstand (DNA-haltig)
-> + 10 ml 100 % EtOH
-> leicht mischen
-> 3-5 min SS34-Rotor (Sorvall) zentrifugieren (austarieren!)
einmal:
-> Überstand abgießen
-> + 10 ml 70 % EtOH
-> zentrifugieren (wie oben)
-> DNA in der "Speed-Vac" (ohne Rotor) oder bei RT trocknen
-> + 150 µl TE-Puffer, Pellet lösen, DNA-Lösung ins
Eppendorfhütchen überführen
-> 5-10 min 65°C (DNA löst sich)
-> + 15 µl 10 mg/ml RNaseA
-> 1/2 h 37°C (RNase-Behandlung, RNA wird abgebaut)
-> 5 min zentrifugieren
-> OD260 nm-Messung:
vorbereiten 4 µl DNA-Lösung + 400 µl H2O
(Messung mit Betreuer/in)
anschl. evtl. NaAc/EtOH-Fällung
-> auf das folgende Test-Agarosegel (siehe Versuch 4) soll
die DNA restringiert und unrestringiert aufgetragen werden
Versuch Nr. 3: 3. Tag (Mittwoch), spätestens 6. Tag (Montag)
Polymerase-Kettenreaktion (PCR) zur Amplifikation des psbA-Gens
Das Prinzip der PCR basiert auf der enzymatischen Amplifikation eines DNA-Bereichs, der von zwei Oligonukleotid-Primern flankiert wird. Die beiden Primer sind so gewählt, daß sie jeweils an einen der beiden DNA-Stränge hybridisieren. Im ersten Reaktionszyklus wird die DNA durch Hitzebehandlung in Einzelstränge zerlegt, anschließend werden die beiden Primer anhybridisiert. In einer durch eine DNA-Polymerase in Gegenwart von dNTPs katalysierten Reaktion werden die Einzelstränge anschließend zum Doppelstrang aufgefüllt. Durch Wiederholung der einzelnen Reaktionsschritte (Denaturierung, Anhybridisierung der Primer, Aufpolymerisierung zum Doppelstrang) erhält man nach 30 Reaktionszyklen eine millionenfache Anreicherung des zwischen den beiden Primern liegenden DNA-Bereiches. Das Verfahren kann (beim Einsatz einer thermostabilen DNA-Polymerase) automatisiert werden.
In diesem Versuch soll der 3'-Bereich des psbA-Gens amplifiziert werden. Das psbA Gen ist das auf der Chloroplasten-DNA lokalisierte Gen für das sog. D1 Reaktionszentrumsprotein von Photosystem II.
Jede Gruppe soll diesen Versuch durchführen, wobei die einzelnen
Gruppenmitglieder getrennte PCR-Ansätze pipettieren können.
(AUFGABE):
Pipettierschema (µl): Handschuhe tragen (Schutz der Proben!), extrem sauber arbeiten!
________________________________________________________________________________________
NK -dNTPs -Primer-Puffer
psbA 3' DNA
_________________________________________________________________________________________
1 2 5 10
DNA (10 ng/µl Klon
388) - 10 10 10 1 2 5 10
Puffer (10x) 5 5 5 -
5 5 5 5
5 mM dNTPs 1 - 1 1 1
1 1 1
T3+-Primer
(50 pmol/µl) 1 1 - 1 1 1 1 1
T7+-Primer (50 pmol/µl)
1 1 - 1 1 1 1 1
H2O ad 50 µl,
mischen!
Thermostabile DNA-Polymerase
1 1 1 1 1 1 1 1
z.B. "PrimeZyme" (2 U/µl)
(mischen!)
NK = "Negativ-Kontrolle" (Welche anderen Kontrollen sind denkbar?)
PCR-Programm Nr. 1 PCR-Programm Nr. 50
(Biometra-Gerät)
30 sec 94 °C
1 min 50 °C 30X 50 °C
1 min 70 °C
Die PCR-Produkte sollen nach der Amplifikation auf einem 6%igen Mini-Polyacrylamidgel getestet werden (von jedem Ansatz: 10 µl PCR-Prod. + 2,5 µl 5x Ficoll-Mix).
Stop-Gel: 3 ml H2O
3 ml Acrylamid-Bisacrylamid 29:1
100 µl 10 % APS
100 µl TEMED
6 % Gel: 5,25 ml H2O
0,75 ml 10X TBE
1,5 ml Acrylamid-Bisacrylamid 29:1
50 µl 10 % APS
50 µl TEMED
Elektrophorese 1 h bei 150 Volt
Nach dem Lauf sollen die PCR-Produkte im Gel durch Ethidiumbromid
und UV-Licht sichtbar gemacht werden (Foto).
pBR/HindIII: 1632, 517, 506, 396, 344, 298, 221 + 220, 154, 75 bp
Versuch Nr. 4: 6. Tag (Montag) bis 8. Tag (Mittwoch)
Southern Analyse der isolierten Gesamt-DNA und der PCR-Produkte
Die isolierte Gesamt-DNA (Versuch 2) und die PCR-Produkte (Versuch 3) sollen zu ihrer Überprüfung nach Restriktion und Gelektrophorese auf Nitrocellulose geblottet und mit einer psbA-Sonde (-> Versuch 5) hybridisiert werden.
Der Southern-Transfer ist ein Verfahren, bei dem DNA-Fragmente durch Gelektrophorese nach ihrer Größe getrennt und anschließend aus der Gelmatrix des Trenngels auf eine geeignete Trägermembran aus Nitrocellulose oder Nylon übertragen und dort immobilisiert werden.
AUFGABE:
Die isolierte Gesamt-DNA soll mit PstI/XbaI und /EcoRI restringiert werden. Pro Ansatz sollen ca. 2-5 µg Gesamt-DNA in einem Volumen von 20 µl mit 10 Units von jedem Restriktionsenzym geschnitten werden(Puffer nicht vergessen, Pipettierschema zur Kontrolle dem Betreuer zeigen). Nach der Restriktion wird die DNA zusammen mit der entsprechenden Menge unrestringierter DNA auf zwei identische große 1 %ige Agarosegele aufgetrennt (20 µl Lambda-Marker nicht vergessen, PCR-Produkte [ca. 10 µl + 2,5 µl 5x Ficoll {außer Kontrollen} ebenfalls auftragen!]). Die Elektrophorese erfolgt über Nacht bei 40 V.
- zweites, unbehandeltes Gel:
- in 0,25 M HCl für 5 min schwenken
(ca. 250 ml aus 1 M HCl ansetzen)
- 2x 15 min in Neutralisationspuffer schütteln
- Gel mit der Handschuhhand beidseitig trocknen (es sollte feucht aber nicht naß sein) und auf die Bögen legen
- Nitrocellulose vorsichtig aus dem Bad nehmen, abblotten und auf das Gel legen
- am nächsten Morgen: Glasscheibe, Papier und Parafilm entfernen,
- Nitrocellulose auf 3 MM Papier 2 h bei 80 °C trocknen.
Zum Nachweis des psbA-Gens soll eine DIG-markierte Sonde verwendet
werden. Digoxigenin wird aus Digitalis purpurea und Digitalis
lanata isoliert. Die Digoxigenin-Markierung eignet sich zur nicht-radioaktiven
Markierung von DNA und RNA. Der Nachweis erfolgt mit Hilfe eines gegen
Digoxigenin gerichteten, spezifischen Antikörpers, der seinerseits
mit einem Enzym gekoppelt ist. Die enzymatische Spaltung eines chromogenen
Substrates durch das am Antikörper gebundene Enzym führt zur
Bildung eines farbigen oder fluoreszierenden Reaktionsproduktes.
Versuch Nr. 5: (nur nach Rücksprache mit den Betreuern durchführen, wenn noch genug Zeit vorhanden; sonst theoretische Behandlung)
Herstellung einer DIG-markierten RNA-Sonde
Die RNA-Sonde wird zur Verfügung gestellt oder neu hergestellt. Die Markierung mit DIG-UTP erfolgt durch in vitro-Transkription eines linearisierten Plasmids (Klon 322/PstI). Der verwendete Vektor (pBluescript, siehe Vektorkarte) trägt den Promoter des Gens für eine phagenkodierte RNA-Polymerase; das hinter diesen Promotor klonierte Gen kann dadurch spezifisch in Form seiner cDNA abgelesen werden.
Vorbereitungen:
linearisierte Template DNA entsprechend restringieren, anschließend Phenol/Choloroform-Behandlung und NaAc/EtOH-Fällung
Markierung:
2) 2 µl 10x Puffer -> Puffer f. Pol
2 µl NTP-Markierungsgemisch (inkl. DIG-UTP)
1 µg DNA
2 µl T3-RNA-Polymerase
ad 20 µl mit Wasser
+ 1 µl RNase-Inhibitor
-> abzentrifugieren
-> 2 h 37°C
-> DNase-Behandlung
-> + 2 µl EDTA-Lösung, 0,2 M pH 8 => Stop
-> 2,5 µl LiCl 4 M
7,5 µl EtOH => RNA-Fällung
-> mind. 30 min -70°C
oder 2 h -20°C
-> 20 min resuspendieren 37 °C
in 50 µl H2O aufnehmen (oder 100 µl), mit 1 µl
RNasin
Versuch Nr. 6: 8. Tag (Mittwoch)
DNA/RNA-Hybridisierung
Die Hybridisierungslösung wird wie im RNA-Teil beschrieben angesetzt (20 ml pro Gruppe) und parallel durchgeführt.
Die Hybridisierungstemperatur beträgt 50°C!
Die Vorhybridisierung sollte mind. 1 h, die Hybridisierung mind. 6 h betragen.
Versuch Nr. 7: 9. Tag (Donnerstag)
DIG-Nachweis
Siehe Versuchsvorschrift im RNA-Teil; der DIG-Nachweis des Southern-
und des Northern-Blots wird zusammen durchgeführt.
II RNA-Versuchsteil (H. Summer)
1. Lösungen
Auflistung in Reihenfolge des Erscheinens in den Versuchsvorschriften; nicht aufgelistet sind ab Werk gelieferte Stammlsg. sowie frisch anzusetzende Lsg. Mit [A] gekennzeichnete Lsg. sind zu autoklavieren.
1.1 TNS-Puffer (Triisopropylnaphtalensulfonsäure) (20 ml)
50 mM Tris HCl pH 7,6
1 % NaCl
2,5 mM MgCl2
2 % TNS (frisch zugeben, lichtempfindlich)
1.2 TNS-gesättigtes Phenol/Chloroform
50 % TNS-gesättigtes Phenol (wird gestellt)
50 % Chloroform
1.3 PstI-Puffer (10x) (10 ml) [A]
60 mM Tris/HCl pH 7,5
500 mM NaCl
60 mM MgCl2
1.4 5 M NH4OAc (50 ml) [A]
1.5 70 % EtOH (50 ml)
1.6 2,5 M NaOAc pH 5 (50 ml) [A]
1.7 10x MOPS-Puffer (MOPS = 4-Morpholinpropan-
sulfonsäure) (200 ml)
200 mM MOPS
50 mM NaOAc
10 mM EDTA
1.8 0,05 M NaOH (1 l) [A]
1.9 0,1 M Tris/HCl pH 8,0 (1 l) [A]
1.10 20x SSC pH 7,0
(mit HCl einstellen am pH-Meter AG-Labor 2/70) (1 l) [A]
3 M NaCL
0,3 M NaCitrat
1.11 Formamid (100 ml) (Vorsicht!)
mit Ionenaustauscher "AG 501-X8(D) mit Indikator" entionisieren (15
min), abfiltrieren und einfrieren
(in Plastikgefäßen mit blauem Deckel
sowie 3 x 1 ml in Eppendorfhütchen)
1.12 H2O bidest. (100 ml) [A]
1.13 10 % N-Laurylsarkosin 100 ml
1.14 20 % SDS 50 ml
1.15 Puffer 1 (Dig-Nachweis) pH 7,5 1 l
Maleinsäure 0,1 M
NaCl 0,15 M
NaOH 5 g
pH mit konz. und verdünnter NaOH bei 20°C einstellen
1.16 Puffer 3 (Dig-Nachweis) pH 9,5 500 ml
Tris/HCl 0,1 M
NaCl 0,1 M
MgCl2 0,05 M
Lösungen einzeln ansetzen, erst dann zusammenfügen
2. Geräte und Materialien
2.1 Eppendorfhütchen (2 ml und 1,5 ml) je in 4 Gläser füllen,
mit Alufolie abdecken -> autoklavieren, danach trocknen
2.2 Mörser, Pistille und Spatel in Alufolie einpacken
und mehrere Stunden bei 180°C sterilisieren
2.3 Pipettenspitzen in Kästen stecken (Handschuhe!),
mit Alufolie bedecken und autoklavieren, danach trocknen
2 Kästen blaue Spitzen
4 Kästen gelbe Spitzen
Allgemeine Sicherheitshinweise: Bei allen Arbeiten mit flüssigem Stickstoff Schutzbrillen tragen. Alle Arbeiten mit Phenol und/oder Chloroform unter dem Abzug durchführen.
- Kotyledonen in vorgekühlten Mörser überführen und in flüssigem Stickstoff zu feinem Pulver (nicht antauen lassen) mörsern, in Puffergemisch aus 4 ml TNS und 5 ml TNS-gesättigtem Phenol/Chloroform in SS34 Röhrchen aus Poylpropylen geben (Röhrchen mit Puffer-Gemisch vorher in flüssigem Stickstoff kurz anfrieren)
- solange schwenken, bis die Mixtur aufgetaut ist, dann auf Eis stellen
- 7 min bei 3.000 rpm bei +4°C in der Sorvall-Zentrifuge im SS34-Rotor zentrifugieren
- Unterphase abziehen (vorsichtig! Vorher üben mit einem Puffer/Lösungsmittel-Gemisch!)
- Ober- und Zwischenphase 1x mit 3 ml TNS-gesättigtem Phenol-Chloroform und 1x mit 5 ml Chloroform reextrahieren
- Oberphase in ein Corexglas überführen, auf Eis stellen
+ 1 Vol 5 M NH4 OAc
+ 1,25 Vol Isopropanol
- 1 - 2 h bei -20°C fällen
- 15 min bei 10.000 rpm bei +4°C (im SS34-Rotor zentrifugieren)
- mit 5 ml 70% EtOH waschen
- im Speed vac trocknen (AG-Labor 2/70) -> (Rotor ausbauen)
- in 360 µl RNase-freiem H2O bidest. aufnehmen
(Sediment soll sich vollständig auflösen)
+ 40 µl 10x Pst-Puffer (RNase-frei!)
+ 15 µl DNase (RNase-frei; 500 µg/ml)
30 min RT, dann auf Eis stellen
- Phenol/Chloroformextraktion mit Phenol/Chloroform - 1/1-Gemisch (v/v), anschließend Ethanolfällung:
+ 1/10 Vol 2,5 M NaOAc pH 5
+ 2 1/2 Vol EtOH
15 min bei -80°C
10 min bei +4°C zentrifugieren (12.000 rpm Eppendorfzentrifuge)
trocknen in Speed vac.
- Sediment in 200 µl RNase-freiem H2O bidest. aufnehmen
- Dreifachbestimmung bei OD260 am Photometer (AG-Labor 2/71)
OD260 x 40 x Verdünnung ergibt µg RNA/ml
Nach der Quantifizierung Proben bei -20°C lagern
Herstellung eines denaturierenden Agarosegels:
Agarosegelkammer mit Aqua bidest. und 100 % EtOH reinigen, mit Klebeband abdichten, Kamm einstellen und unter den Abzug stellen.
- 1,7%ige Agarose aufkochen und auf ca. 50°C abkühlen lassen (z.B. in Behälter mit warmem Wasser). MOPS-Puffer und Formaldehyd zugeben, wenn die Agaroselsg. etwa handwarm ist, mischen und Lösung blasenfrei in die Gelkammer gießen
(Rest an Formaldehyd für den Probenpuffer aufbewahren)
- Elektrophorese-Apparatur mit 1x MOPS-Puffer füllen und das Gel einlegen, wenn es erstarrt ist
H2O 39 ml
Agarose 0,66 g
10x MOPS-Puffer 6 ml
Formaldehyd 11,25 ml
Herstellung und Auftrennung der RNA-Proben
Lösungen (frisch ansetzen):
1) Probenpuffer:
(ca. 1 h haltbar)
Stamm für 10 Spuren für 50 Spuren
H2O 28 µl 140 µl
100 % Formamid 50 µl 250 µl
Formaldehyd 17 µl 85 µl
10x MOPS-Puffer 5 µl 25 µl
2) Gelbeladungspuffer:
Stamm für 10 Spuren für 50 Spuren
100 % Formamid 25 µl 50 µl
100 % Glycerin 20 µl 40 µl
2 % BPB (Bromphenolblau)* 2,5 µl 5 µl
2 % XCFF (Xylencyanolblau)* 2,5 µl 5 µl
* werden gestellt
- 10 µl Probenpuffer zugeben, whirlen und abzentrifugieren
Probe 10 min bei 65 °C im Wasserbad inkubieren,
sofort auf Eis stellen
- 2 µl Gelbeladungspuffer zugeben
- Probe auf Gel auftragen
- Elektrophorese, 1-2 h bei 80 Volt
Herstellung der "Blots" (Transferfolien)
Nach dem Elektrophoreselauf wird das Gel in folgenden Lsg. geschwenkt:
10 min in H2O
15 min in Ethidiumbromidlsg. (100 µg/ml) (nicht beim DIG-Nachweis)
10 min in 0,05 M NaOH schütteln
20 min in 0,1 M Tris/HCl pH 8,0 schütteln
(evtl. Polaroidfoto am Transilluminator AG-Labor 2/73)
30 min in 10x SSC schütteln
2) 2 Streifen 3MM-Papier in 10x SSC einweichen und über eine Glassscheibe legen, so daß die Enden in die Flüssigkeit eintauchen
3) abgestrichenes Gel darauf legen
4) Nitrocellulose erst in H2O benetzen, dann 20x SSC zugeben (Endkonz. 10x SSC) und luftblasenfrei auf das Gel legen (in der Mitte zuerst!)
5) überstehende Nitrocellulose mit Parafilm abdecken
6) 2x 3MM-Papier in 10x SSC anfeuchten und auf die Nitrocellulose legen
7) ca. 6 Lagen trockenes 3MM-Papier auflegen und einen Stapel Saugpapier (z.B. ungebleichte Cellulose) darüberlegen
8) hierauf eine Glasscheibe legen und
mit einem Gewicht (Flasche) beschweren, über Nacht (mind. 12 h) blotten
4. Tag
- Blot-Vorrichtung auseinanderbauen und die Gelumrisse sowie die Taschen und die Orientierung auf der Nitrocellulose markieren
- Nitrocellulose auf den Transilluminator legen und die ribosomalen RNAs markieren
- Nitrocellulose zwischen 2 Lagen 3MM-Papier
trocknen lassen, 2 h bei 80°C "backen" (Hitzefixierung!), Blot danach
in Folie einschweißen und anschließend bei +4°C lagern.
2. Woche/1. Tag
RNA/RNA-Hybridisierung mit Digoxigenin-markierter DNA-Sonde
Vorhybridisierung: Filter aus der Folie nehmen, in eine Hybridisierungsröhre geben und ca. 1 h mit 20 ml Vorhybridisierungspuffer/100 cm2 Filterfläche bei 68°C inkubieren. Es sollten keine Luftblasen zwischen Wandung und Filter sichtbar sein. Den Filter nur mit gereinigten Pinzetten berühren.
* wird gestellt
Hybridisierung:
2. Tag/2. Woche
50 ml/100 cm2 Filterfläche
- 2 x 15 min bei 68°C 0,1 x SSC/0,1 % SDS
50 ml/100 cm2 Filterfläche
Die Waschschritte können direkt in der Glasröhre erfolgen.
Chemilumineszenz-Nachweisreaktion zur Detektion hybridisierter RNA
Lösungen für 100 cm2 Membran
Puffer 3: Tris-HCl 0,1 mol/l; NaCl 0,1 mol/l; MgCl2 50 mmol/l
pH 9,5 (20°C)
Puffer 4: 1 % Magermilchpulver in Puffer 1
- Membranen kurz waschen (1-5 min) in Wasch-Puffer
(= Puffer 1 + 0,3 % Tween 20) ca. 200 ml
- 30' min in 100 ml Puffer 4
- Anti-DIG-AP-Konjugat auf 75 mU/ml (1:10.000) in Puffer 2
verdünnen (Verdünnung 12 h bei 4°C stabil)
(2,0 µl + 20 ml Puffer 2)
- Filter 30 min in 20 ml verdünnter Antikörper-Konjugatlsg.
inkubieren (Inkubation in Plastikschale)
- 2 x 15 min mit 100 ml Waschpuffer waschen
verdünnen
(5x wiederverwendbar, bei 4°C im Dunkeln lagern)
- Überschüssige Flüssigkeit von der Membran ablaufen lassen
- Feuchte Membran auf Plastikhalter legen, mit Frischhaltefolie
"einkleiden" und 5-15 min bei 37°C präinkubieren Brutschrank) - Röntgenfilm mehrere Stunden bei RT auflegen (in Dunkelkammer
Arb.gruppen-Labor 2/67) Röntgenfilm entwickeln, bei zufrieden
stellenden Resultaten die Membran feucht einschweißen
(evtl. Rehybridisierung)
* wird gestellt
Alle Schritte werden bei Raumtemperatur unter Schütteln durchgeführt (in kleinen Plastikschalen).
Entfernen von DIG-markierter DNA zur Rehybridisierung von Southern-/Northern- oder Dot-Blots
(wird nur durchgeführt, wenn genügend Zeit vorhanden ist)
- Die Membran in einer Lösung mit 50 mM EDTA, pH 8,0, 2 x SSC bei 85°C für 15 min inkubieren.
- zweimal in 2 x SSC, 0,1 % (w/v) SDS bei 37°C für 5 min waschen.
- zweimal in 0,1 % (w/v) SDS, 0,05 N NaOH bei 37°C für 15 min inkubieren.
- Membran mit 2 x SSC spülen.
Lösungen:
Aufschlußpuffer 2 l Endkonz.
Saccharose 342,2 g 0,5 M
2M Tris/HCl pH 8 100 ml 0,1 M
1M MgCL2 20 ml 0,01 M
BSA 2 g 0,1 %
Mercaptoethanol 5,76 ml 0,04 M
Gradientenlösungen 150 ml Endkonz
20 % Saccharose 30 g 20 % (w/v)
2M Tris/HCl pH 8 3,75 ml 0,05 M
1M MgCL2 1,5 ml 0,01 M
Mercaptoethanol 0,432 ml 0,04 M
BSA 0,15 g 1 %
mit H2O auf 150 ml auffüllen
150 g Endkonz.
60 % Saccharose 90 g 60 % (w/w)
2M Tris/HCl pH 8 3,75 ml 0,05 M
1M MgCL2 1,5 ml 0,01 M
Mercaptoethanol 0,432 ml 0,04 M
BSA 0,15 g 1 %
mit H2O auf 150 g auffüllen
Lösungen für Chlorophyllbestimmung
80 % Aceton
Lösungen für Proteinnachweis nach Bradford
Bradford-Lösung
65 mg Serva-Blau G (= Coomassie Brilliant Blue G) in 50 ml EtOH lösen (Farbstoff: Bitte sauber und vorsichtig handhaben!), anschließend 135 ml 85 % Phosphorsäure dazugeben (Vorsicht!!!)
und mit H2O auf 200 ml auffüllen
Lösung wird bei 4°C aufbewahrt.
Lösungen für SDS-Gelelektrophorese
Endkonzentration
H2O 20 ml
2. Trenngel: (30 ml pro Gel)
Acrylamid/Bis. (30 %/0,8 %) 12,5 %
12,5 ml
Tris/HCl pH 8,8 0,5 % 10 ml
SDS 0,1 % 170 µl reicht für
APS 0,02% 30 µl 1 Gel
TEMED 0,2 % 30 µl
H2O 2,5ml
3. Elektrolytlösung (600 ml per
Gel)
Tris 0,3 %
Glycin 1,4 %
SDS 0,1 %
4. SDS-Probenpuffer
Tris/HCl pH 6,8 60 mM
ß-Mercaptoethanol 5 %
SDS 2 %
Bromphenolblau 0,04 %
5. Färbe-Lösung
Methanol 50 %
Essigsäure 10 %
Commassie brilliant blue R250 0,4
%
(Serva)
6. Entfärbe-Lösung
Methanol 20 %
Essigsäure 10 %
Achtung! Das momomere Acrylamid
ist krebserregend. Spritzer auf Haut oder in die Augen (Schutzbrille) mit
Wasser sofort abspülen. Handschuhe!
Geräte:
Plastidenisolierung:
1 Waring Blendor, 200 ml Meßzylinder, 2 1l-Bottiche, 4 GS3 Becher, 1 Pinsel, 1 großer Trichter, 1 Potter, 1 Glaspipette (20 ml), GS3 Rotor, UZ-Rotor TST 28.38, UZ Rotor Buckets, 1 großes Eisbad, Alufolie, 4 Lagen dopp. Verbandmull, 2 Lagen 4-fach Verbandmull, 2 Lagen Miracloth
=> kalt stellen
1 1-ml-Pipette, 40 Reagenzgläser oder 2-ml-Eppendorfgefäße
Chlorophyllbestimmung:
Glasküvetten, 1 Photometer
Proteinbestimmung nach Bradford:
Bechergläser, 1 Glasfilter, Filterpapier, 1 Photometer
SDS-Gelelektrophorese:
2 Gelkammern, 4 mittelgroße Glasplatten, 4 Spacer,
1 200 µl-Pipette
gelbe und blaue Eppendorfspitzen
_________________________
Organell-Isolierung - Proteine
1. Woche/1. Tag Pflanzenaussaat Licht/Dunkel-Pflanzen
(Erde / Vermiculit)
Jeweils zwei Anzuchtschalen mit Blumenerde bzw. Vermiculit (morgens vorquellen) zu etwa 2/3 (Vol) ausfüllen. Schalen mit Blumenerde in der Phytokammer gießen und Erde "kneten", so daß sie nicht nur oberflächlich befeuchtet ist (jedoch nicht zu feucht!); Senfsamen so dicht aussäen, daß sich eine nahezu geschlossene, aber einlagige Samendecke ergibt. Erneut vorsichtig gießen und Senfsamen nochmals vorsichtig andrücken.
Samen auf Vermiculit etwas weniger
dicht aussäen, nochmal gießen, leicht andrücken und Pflanzenschalen
umgehend in den Dunkelanzuchtraum stellen.
1. Woche/ 3. Tag
Ansetzen der Lösungen siehe oben
Kaltstellen der Geräte siehe oben
Gießen der Gradienten
Herstellen von linearen Gradienten
In Gradientenröhrchen jeweils ein 2 ml-"Kissen" der 60 % (w/w) Saccharoselsg. gießen. Anschließend den linearen Gradienten darüberschichten. 15 ml 30 % (w/v) und 15 ml 60 % (w/v) Saccharoselsg. in eine Mischkammer geben und vorsichtig auf das Kissen schichten. Gradienten im Kühlraum aufbewahren.
1. Woche/ 5. Tag
Die Arbeiten erfolgen parallel in zwei Gruppen!
Eine Gruppe präpariert Etioplasten, die andere Chloroplasten. Die Arbeiten für die Isolierung beider Plastidentypen sind identisch, mit der Ausnahme, daß die Etioplastenpräparation bis zum ersten Zentrifugationsschritt unter grünem Sicherheitslicht stattfindet. Alle Arbeiten finden bei 4°C statt, die Zentrifugationsschritte laufen parallel. (Plastiden auf allen Reinigungsschritten kalt halten: Proteasen!)
Der Arbeitsablauf grob skizziert:
- Homogenisation des Pflanzenmaterials
-> mechanischer Zellaufschluß
- Reduktion des Volumens = Konzentrierung des Homogenates
-> differentielle Zentrifugation
- Vereinzeln der Plastiden
- Dichtegradientenzentrifugation zur Auftrennung
der Zellbestandteile
Jeweils 200 g Senfkotyledonen abpräparieren (Handschuhe; auf Eis). Hiervon jeweils ca. 50 g mit 200 ml Aufschlußpuffer im Waring Blendor homogenisieren (2 x 6 sec low + 1 x 6 sec high). Homogenat von 100 g Kotyledonen durch eine Lage Verbandmull pressen. Diesen Schritt wiederholen. Gesamtes Homogenat durch eine doppelte Lage Mull und anschließend durch eine doppelte Lage Miracloth filtrieren. Filtrat auf 2GS3 Becher verteilen und 15 min bei 7500 rpm im GS3 Rotor zentrifugieren.
Überstand vorsichtig über das Pellet abgießen. "Pellet" (Sediment) mit einem Pinsel VORSICHTIG resuspendieren und Resuspendat auf etwa 18 ml Gesamtvolumen mit Puffer A auffüllen (Becher auf Eis halten!!!).
Da Plastiden nach dieser Behandlung aggregieren, müssen die Aggregate durch VORSICHTIGES Pottern (nur unter Anleitung!) vereinzelt werden. Nach dieser Behandlung können nun 8 ml der Suspension auf die Saccharose-Dichtegradienten geschichtet werden. Anschließend Rotor und Zentrifuge unter ANLEITUNG beladen und bei 23.000 rpm 45 min lang zentrifugieren.
Nach erfolgter Zentrifugation wird
der Gradient in 20 Fraktionen à 2 ml zerlegt (ANLEITUNG).
Mit den einzelnen Fraktionen werden
jeweils folgende Tests durchgeführt:
Chlorophyllbestimmung: (Gesamtchlorophyll in den Fraktionen des Saccharosegradienten).
0,05 ml Probe + 2,45 ml 80 % Aceton mischen, ggf. zentrifugieren.
Extinktion bei 652 nm gegen 80 % Aceton messen.
E652 x 1,45 = mg Chlorophyll/ml Probe. (Glasküvetten)
2. Woche/ 3. + 4. Tag
Proteinbestimmung:
0,1 ml einer Proteinlösung werden mit 1,0 ml Bradford-Verdünnung versetzt und vermischt. Nach 30 min kann die Extinktion bei 595 nm gegen den Leerwert (100 µl Puffer statt Probe + 1 ml Bradford-Verdünnung) gemessen werden.
Zunächst wird eine Eichkurve mit Rinderserum albumin angefertigt (0; 2; 4; 6; 8; 10; 20; 30; ...... 60 µg BSA/100 µl Probe) (Dreichfachbestimmung!).
Nach der Erstellung der Eichkurve werden jeweils 100 µl der zum Teil verdünnten Gradientenfraktionen mit 1 ml Bradford-Verdünnung versetzt (vgl. 2b) (Doppelbestimmungen).
(Fraktionen 1-6 1:20 verdünnen;
Fraktionen 7-12 1:5 verdünnen; Fraktionen 13-19 unverdünnt, letzte
Fraktion 1:3 verdünnen). Die erhaltenen Meßwerte mit der Eichkurve
vergleichen und die Verdünnungen eventuell anpassen. Aus den Meßwerten,
die im linearen Bereich der Eichkurve liegen, wird der Proteingehalt der
Fraktionen ermittelt (Verdünnungen berücksichtigen!)
SDS-Gelelektrophorese:
Proteine verfügen an ihrer Oberfläche in der Regel über eine negative oder positive Netto-Ladung, die vom Gehalt an sauren (Glutamin- oder Asparaginsäure) und basischen Aminosäuren (Lysin und Arginin) abhängt. In einer Proteinlösung, an die ein elektrisches Feld angelegt wird, werden die Proteine entsprechend dieser Nettoladung entweder zur Kathode oder Anode wandern. Die Wanderungsgeschwindigkeit der Proteine hängt dabei von der elektrischen Feldstärke, der Viskosität des Mediums sowie von Größe und Gestalt des Proteins selber ab. Dieses Prinzip der Auftrennung nennt man Elektrophorese.
Üblicherweise trennt man Proteine über ein Gel auf, das eine inerte Matrix darstellt. Diese Matrix setzt die Durchmischung der Proteinprobe herab, ohne die Wanderungsgeschwindigkeit der Proteine zu beeinflussen. Die Matrix entsteht durch Polymerisation von monomerem Acrylamid. Durch ein Quervernetzungsreagenz, dem N,N'-Methylen-bisacrylamid, werden die Acrylamidketten in Gegenwart eines Katalysators zu einem Gel quervernetzt. Der Gehalt an Bisacrylamid bestimmt dabei die Porengröße des Gels und somit die Proteinauftrennung.
Damit die Proteine in einem Gleichspannungsfeld alle in dieselbe Richtung wandern, hebt man die individuellen Nettoladungen durch Behandlung mit einem stark negativ geladenen Detergens auf. Der Dodecylsulfatrest des Natrium-(Sodium)-dodecylsulfats (SDS) interkaliert zwischen den hydrophoben Aminosäureresten von "Proteinkernen", so daß alle Protein-SDS- Aggregate eine negative Nettoladung erhalten. Eventuell vorhandene Disulfidbrücken zu anderen Polypeptiden können durch Behandlung mit ß-Mercaptoethanol, einer stark reduzierend wirkenden Substanz, gespalten werden. Die Proteine wandern nun bei Anlegen einer Gleichspannung an die poröse Matrix des Gels zur positiven Elektrode und trennen sich dabei entsprechend ihrer Größe (Molekulargewicht) voneinander, d.h. kleine Proteine wandern schneller als große. Durch Vergleich mit Proteinen bekannter Größe (sog. Markerproteine) lassen sich die im Gemisch der Probe enthaltenen Proteine nach erfolgter Auftrennung und Anfärbung auf ihre Größe hin analysieren.
Bei der hier durchgeführten SDS-PAGE
handelt es sich um das diskontinuierliche Gelsystem nach Laemmli (1970),
dem gebräuchlichsten Gelsystem. In einem Sammelgel werden die in Taschen
aufgetragenen Proteinproben zunächst zu einer scharfen Bande konzentriert
und in einem nachfolgenden Trenngel nach ihrer Größe aufgetrennt.
Durch Zugabe eines Farbmarkers in die Proteinprobe, z.B. Bromphenolblau,
läßt sich die Auftrennung optisch verfolgen. Nach Beendigung
des Gellaufes werden die Proteine in der Gelmatrix fixiert und angefärbt.
2. Das Gel wird am nächsten Morgen aus dem Elektrophorese-Apparat entfernt und in einer Glasschale mit Färbe-Lösung bedeckt. Auf einem Schüttler wird das Gel bei leichtem Schütteln 20 min gefärbt.
3. Die Färbelösung wird abdekantiert und das Gel in Entfärberlösung weitergeschüttelt, bis die Banden sichtbar werden. Ein eingetauchtes KleenexR-Tuch erhöht die Entfärbe-Geschwindigkeit.
4. Nach erfolgter Entfärbung werden
die Polypeptidspektren entlang der Saccharosegradienten von Licht- und
Dunkelpflanzen verglichen. Durch Vergleich mit den 'Markerbanden' werden
die Molekulargewichte charakteristischer Polypeptidbanden bestimmt und
den verschiedenen Organelltypen zugeordnet.
Ibelgaufts, H. (1992). "Gentechnologie von A-Z", VHC.
Maliga, P., Klessig, D.F., Cashmore, A.R., Gruissem, W. und Varner, J.E. (1995). "Methods in Plant Molecular Biology - A Laboratory Course Manual", Cold Spring Harbor Laboratory Press.
Sul, I-W. und Korban, S.S. (1996). A highly efficient method for isolating genomic DNA from plant tissues. Plant Tissue Culture and Biotechnology Vol. 2, 113-116.
Richter, G. (1996). "Biochemie der Pflanzen", Thieme-Verlag.
Westhoff, P., Jeske, H. Jürgens,
G., Kloppstech, K. und Link, G. (1996). "Molekulare Entwicklungsbiologie
- Vom Gen zur Pflanze", Thieme-Verlag.
Auswahl wichtiger Methoden der modernen Molekularbiologie
10 (bis max. 15) min pro Vortrag!
Ziele: methodisches Prinzip; besondere Schwierigkeiten u. Tips
& Tricks zur Überwindung; Anwendungszweck mit Blick auf pflanzliche
Systeme
Seminarthemen:
1. Prinzipien der DNA-Klonierung
2. DNA-Sequenzierung nach Sanger
3. Anlage einer cDNA-Bank
4. "Western-/Immunoblotting"
5. "Differential Display"
6. "Two-Hybrid"-System
7. Protein-Sequenzierung
8. Datenbank-Recherche via Internet
Empfohlene Literatur:
Bertram und Gassen 1991: "Gentechnische Methoden" Gustav Fischer Verlag
Gassen, Martin, Bertram 1987: "Gentechnik" (2.Auflage) Gustav Fischer
Verlag
Sambrook et al. 1989: "Molecular Cloning" (2.Edition) Cold Spring Harbor
Laboratory Press
Ausubel et al. (ständig erneuert): "Current Protocols in Molecular
Biology" Wiley Interscience