
| Endocrine Index | Glossary |
|---|
|
Physiologic Effects of Insulin |
|
Stand on a streetcorner and ask people if they know what insulin is, and many will reply, "Doesn't it have something to do with blood sugar?" Indeed, that is correct, but such a response is a bit like saying "Mozart? Wasn't he some kind of a musician?" Insulin is a key player in the control of intermediary metabolism. It has profound effects on both carbohydrate and lipid metabolism, and significant influences on protein and mineral metabolism. Consequently, derangements in insulin signalling have widespread and devastating effects on many organs and tissues. The Insulin Receptor and Mechanism of ActionLike the receptors for other protein hormones, the receptor for insulin is embedded in the plasma membrane. The insulin receptor is composed of two alpha subunits and two beta subunits linked by disulfide bonds. The alpha chains are entirely extracellular and house insulin binding domains, while the linked beta chains penetrate through the plasma membrane. 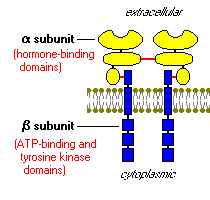
The insulin receptor is a tyrosine kinase. In other words, it functions as an enzyme that transfers phosphate groups from ATP to tyrosine residues on intracellular target proteins. Binding of insulin to the alpha subunits causes the beta subunits to phosphorylate themselves (autophosphorylation), thus activating the catalytic activity of the receptor. The activated receptor then phosphorylates a number of intracellular proteins, which in turn alters their activity, thereby generating a biological response. Several intracellular proteins have been identified as phosphorylation substrates for the insulin receptor, the best-studied of which is insulin receptor substrate 1 or IRS-1. When IRS-1 is activated by phosphorylation, a lot of things happen. Among other things, IRS-1 serves as a type of docking center for recruitment and activation of other enzymes that ultimately mediate insulin's effects. A more detailed look at these processes is presented in the section on Insulin Signal Transduction. Insulin and Carbohydrate MetabolismGlucose is liberated from dietary carbohydrate such as starch or sucrose by hydrolysis within the small intestine, and is then absorbed into the blood. Elevated concentrations of glucose in blood stimulate release of insulin, and insulin acts on cells thoughout the body to stimulate uptake, utilization and storage of glucose. The effects of insulin on glucose metabolism vary depending on the target tissue. Two important effects are: |
|
1. Insulin facilitates entry of glucose into muscle, adipose and several other tissues. The only mechanism by which cells can take up glucose is by facilitated diffusion through a family of hexose transporters. In many tissues - muscle being a prime example - the major transporter used for uptake of glucose (called GLUT4) is made available in the plasma membrane through the action of insulin. | |
|
In the absense of insulin, GLUT4 glucose transporters are present in cytoplasmic vesicles, where they are useless for transporting glucose. Binding of insulin to receptors on such cells leads rapidly to fusion of those vesicles with the plasma membrane and insertion of the glucose transporters, thereby giving the cell an ability to efficiently take up glucose. When blood levels of insulin decrease and insulin receptors are no longer occupied, the glucose transporters are recycled back into the cytoplasm. The animation to the right depicts how insulin signalling leads to translocation of glucose transporters from the cytoplasm into the plasma membrane, allowing glucose (small blue balls) to enter the cell. Click on the "Add Glucose" button to start it. |
|
|
It should be noted here that there are some tissues that do not require insulin for efficient uptake of glucose: important examples are brain and the liver. This is because these cells don't use GLUT4 for importing glucose, but rather, another transporter that is not insulin-dependent. 
2. Insulin stimulates the liver to store glucose in the form of glycogen. A large fraction of glucose absorbed from the small intestine is immediately taken up by hepatocytes, which convert it into the storage polymer glycogen. Insulin has several effects in liver which stimulate glycogen synthesis. First, it activates the enzyme hexokinase, which phosphorylates glucose, trapping it within the cell. Coincidently, insulin acts to inhibit the activity of glucose-6-phosphatase. Insulin also activates several of the enzymes that are directly involved in glycogen synthesis, including phosphofructokinase and glycogen synthase. The net effect is clear: when the supply of glucose is abundant, insulin "tells" the liver to bank as much of it as possible for use later. | |
|
A well-known effect of insulin is to decrease the concentration of glucose in blood, which should make sense considering the mechanisms described above. Another important consideration is that, as blood glucose concentrations fall, insulin secretion ceases. In the absense of insulin, a bulk of the cells in the body become unable to take up glucose, and begin a switch to using alternative fuels like fatty acids for energy. Neurons, however, require a constant supply of glucose, which in the short term, is provided from glycogen reserves. In the absense of insulin, glycogen synthesis in the liver ceases and enzymes responsible for breakdown of glycogen become active. Glycogen breakdown is stimulated not only by the absense of insulin but by the presence of glucagon, which is secreted when blood glucose levels fall below the normal range. Insulin and Lipid MetabolismThe metabolic pathways for utilization of fats and carbohydrates are deeply and intricately intertwined. Considering insulin's profound effects on carbohydrate metabolism, it stands to reason that insulin also has important effects on lipid metabolism. Important effects of insulin on lipid metabolism include the following: |
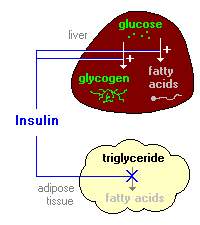
Insulin promotes synthesis of fatty acids in the liver. As discussed above, insulin is stimulatory to synthesis of glycogen in the liver. However, as glycogen accumulates to high levels (roughly 5% of liver mass), further synthesis is strongly suppressed. When the liver is saturated with glycogen, any additional glucose taken up by hepatocytes is shunted into pathways leading to synthesis of fatty acids, which are exported from the liver as lipoproteins. The lipoproteins are ripped apart in the circulation, providing free fatty acids for use in other tissues, including adipocytes, which use them to synthesize triglyceride. Insulin inhibits breakdown of fat in adipose tissue by inhibiting the intracellular lipase that hydrolyzes triglycerides to release fatty acids. Insulin facilitates entry of glucose into adipocytes, and within those cells, glucose can be used to synthesize glycerol. This glycerol, along with the fatty acids delivered from the liver, are used to synthesize triglyceride within the adipocyte. By these mechanisms, insulin is involved in further accumulation of triglyceride in fat cells. From a whole body perspective, insulin has a fat-sparing effect. Not only does it drive most cells to preferentially oxidize carbohydrates instead of fatty acids for energy, insulin indirectly stimulates accumulation of fat is adipose tissue. |
Other Notable Effects of InsulinIn addition to insulin's effect on entry of glucose into cells, it also stimulates the uptake of amino acids, again contributing to its overall anabolic effect. When insulin levels are low, as in the fasting state, the balance is pushed toward intracellular protein degradation. Insulin also increases the permiability of many cells to potassium, magnesium and phosphate ions. The effect on potassium is clinically important. Insulin activates sodium-potassium ATPases in many cells, causing a flux of potassium into cells. Under certain circumstances, injection of insulin can kill patients because of its ability to acutely suppress plasma potassium concentrations. Insulin Deficiency and Excess DiseasesDiabetes mellitus, arguably the most important metabolic disease of man, is an insulin deficiency state. It also is a significant cause of disease in dogs and cats. Two principal forms of this disease are recognized:
Hyperinsulinemia or excessive insulin secretion is usually the result of an insulin-secreting tumor. This condition is much less common than diabetes mellitus. The high levels of insulin resulting from this condition or from an overdose of insulin causes a precipitious drop in blood glucose concentrations. The brain becomes starved for energy, leading to the syndrome of insulin shock, which is acutely life-threatening. |
| Index of: The Endocrine Pancreas | |||
|---|---|---|---|
 |
Insulin Synthesis and Secretion | Glucagon |  |
|
Last updated on October 17, 2004 |
| Author: R. Bowen |
| Send comments via form or email to rbowen@colostate.edu |