Diese Seite gibt eine kurze
Einführung in die verschiedenen Forschungsprojekte der Projektgruppe "Functional Genomics-Analysen der Morphogenese von Pilzen".
Detaillierte Informationen hierzu gibt es in mehreren
Veröffentlichungen. Interessierte
Studierende können im Rahmen von S-Modulen am
Lehrstuhl für Molekulare und Zelluläre Botanik an aktuellen Forschungsthemen mitarbeiten. Diese vier- oder sechs-wöchigen Laborpraktika können
zu Bachelor- oder Masterarbeitsprojekten hinführen.
Vielzellige Entwicklung, wie z.B. die Entwicklung von
Pilzfruchtkörpern, kann nur korrekt ablaufen, wenn viele Gene koordiniert exprimiert werden. Die Gene müssen zur richtigen
Zeit und am richtigen Ort (d.h. in den richtigen Zellen des Organismus) aktiviert oder deaktiviert werden, um einen
funktionellen Fruchtkörper zu ergeben. Auch Signale von Außen, wie z.B. Temperatur, Feuchtigkeit, Nährstoffangebot und viele
andere müssen berücksichtigt werden. Während der letzten Jahre wurden Methoden wie Mikroarrays und RNA-Seq entwickelt, mit denen die
Expression von hunderten oder tausenden von Genen gleichzeitig untersucht werden kann. Im Moment arbeiten wir an Projekten
zur Analyse der differentiellen Genexpression bei verschiedenen Hyphenpilzen sowie an "comparative genomics"-Projekten.
Untersuchungen wie diese liefern Informationen darüber, wie ein Pilzgenom vielzellige Entwicklung kontrolliert.
In einem zweiten Bereich unserer Forschung untersuchen wir die Evolution der Kreuzungstyp-Loci in Basidiomyceten. Kreuzungstyp-Loci entscheiden, welche Stämme miteinander kompatibel für die
sexuelle Entwicklung sind.
1. "Functional genomics"-Analysen der Fruchtkörperentwicklung bei dem Hyphenpilz Sordaria macrospora
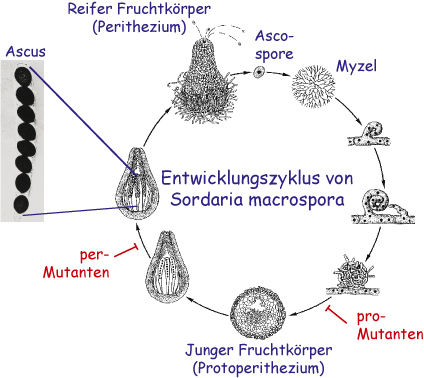
Sordaria macrospora ist ein Hyphenpilz, der sehr nahe mit Neurospora crassa verwandt ist. Aber im Gegensatz zu Neurospora ist Sordaria homothallisch, was bedeutet, dass ein einzelner Stamm Fruchtkörper (Perithezien) bilden kann, ohne dafür einen Partner von entgegengesetztem Kreuzungstyp zu benötigen. Der Entwicklungszyklus von S. macrospora ist links abgebildet. Er beginnt mit einer Ascospore, die auskeimt und zu einem Myzel auswächst. Innerhalb von einer Woche werden Perithezien gebildet, in denen Asci produziert werden, die jeweils acht Ascosporen enthalten. Die Ascosporen werden aus dem Fruchtkörper ausgeschleudert und der Zyklus beginnt von Neuem.
- Bei mehreren Sordaria-Mutanten ist der Entwicklungszyklus an einem bestimmten Punkt unterbrochen, so dass keine reifen Sporen mehr gebildet werden. Einige dieser Mutanten bilden nur sogenannte Vorfruchtkörper (Protoperithezien), daher werden sie pro-Mutanten genannt. Mutanten, die zwar Fruchtkörper (Perithezien) produzieren, aber keine reifen Sporen, werden als per-Mutanten bezeichnet.
- Ein Fokus unserer Analysen liegt auf der Rolle von Chromatin-Modifiern und Transkriptionsfaktoren bei der vielzelligen Entwicklung. In früheren Untersuchungen konnten wir zeigen, dass das Histon-Chaperon ASF1 und der Transkriptionsfaktor PRO44 essentiell für die Fruchtkörperentwicklung in Sordaria macrospora sind. In einer kürzlich durchgeführten Studie haben wir nun die Funktion der Entwicklungsgene asf1 und pro44 in Sordaria macrospora mittels RNA-seq, MNase-seq, BS-seq, Fluoreszenz-Mikroskopie und Protein-Protein-Interaktionsstudien detailliert untersucht. Unsere Untersuchungen zeigen, dass die beiden Gene verschiedene Aspekte der sexuellen Entwicklung regulieren, obwohl sie im gleichen Stadium der Fruchtkörperentwicklung benötigt werden. Während asf1 als Suppressor von schwach exprmierten Genen während der Morphogenese agiert, zeigten Transkriptom-Analysen in jungen Fruchtkörpern, dass pro44 für die korrekte Expression von Genen benötigt wird, die in den extrazellulären Metabolismus involviert sind. Die Deletion des putativen Transkriptionsfaktor-Gens asm2, das in jungen Fruchtkörpern der pro44 Mutante herunterreguliert ist, führte zu Defekten in der Ascosporen-Bildung. Mehr Informationen gibt es in unserer Veröffentlichung dieser Studie (Schumacher et al. 2018, BMC Genetics 19: 112).
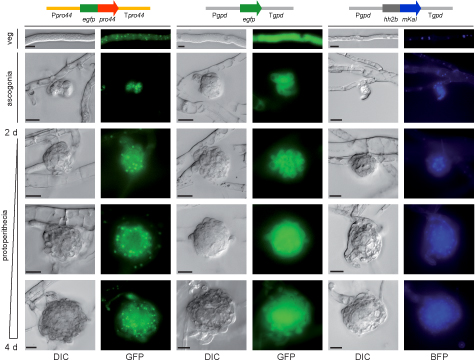
Die Abbildung zeigt die Lokalisation des PRO44-Proteins in sich entwickelnden Fruchtkörpern. PRO44 wurde mit dem grün-fluoreszierenden Protein (GFP) fusioniert, um die Lokalisation mit Fluoreszenzmikroskopie zu analysieren. Das Fusionskonstrukt wurde unter Kontrolle der pro44-Promotor- und -Terminator-Region exprimiert (links). Als Kontrolle wurde ein Konstrukt verwendet, das nur GFP exprimiert (Mitte), sowie ein Fusionsprotein aus einem blau-fluoreszierenden Protein und dem Histon H2A, das im Zellkern lokalisiert (rechts). PRO44 befindet sich im Zellkern, in sich entwickelnden Fruchtkörpern allerdings vorwiegend in den äusseren Schichten und nicht im Inneren des Fruchtkörpers.
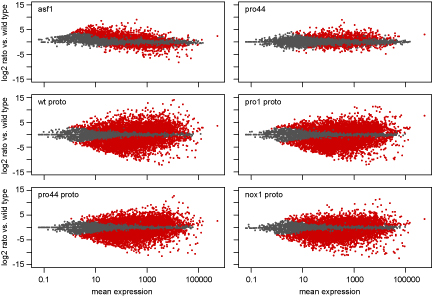
Als Teil dieser Untersuchungen haben wir eine Kombination aus Laser-Mikrodissektion (LM) und RNA-seq verwendet. LM ermöglicht es, junge Fruchtkörper getrennt vom umgebenden vegeativen Myzel zu isolieren. Wir können RNA aus diesen Fruchtkörpern extrahieren und für RNA-seq verwenden. Die Abbildung zeigt einen MA-plot, bei dem die Expression für alle Gene die Expression des Gens in einem bestimmten Vergleich (z.B. Mutante gegen Wildtyp) gegen die durchschnittliche Expression des Gens in allen untersuchten Bedingungen aufgetragen ist. Gene, die in rot dargestellt sind, sind im analysierten Vergleich signifikant hoch- oder herunterreguliert.

Eines der Gene, das in der pro44-Mutante anders reguliert ist als im Wildtyp, ist asm2. Das Gen kodiert für einen putativen Transkriptionsfaktor, und seine Deletion führt zu Defekten in der Ascosporenreifung, die in der linkz gezeigten Abbildung zu sehen sind. Die Mutante und der Wildtyp wurden für ein bis zwei Wochen auf verschiedenen Medien (BMM und SWG) angezogen. Ein komplementierter Stamm (dritte Zeile für beide Zeitpunkte) hat wieder den Wildtyp-Phänotyp.
2. Vergleichende Genexpressionsanalysen ("comparative functional genomics") mit Sordaria macrospora, Neurospora crassa und Pyronema confluens
- Die Fruchtkörper von filamentösen Ascomyceten wie S. macrospora, N. crassa und P. confluens sind wahrscheinlich homologe Strukturen, d.h. sie sind aus einem gemeinsamen Vorfahr entstanden. Daher ist es wahrscheinlich, dass die Genexpressionsmuster, die für der Fruchtkörperentwicklung nötig sind, ebenfalls zu einem gewissen Grad konserviert sind. Um mehr darüber zu erfahren, welche Gene während der Fruchtkörperentwicklung in verschiedenen Pilz-Spezies exprimiert werden, haben wir ein Projekt begonnen, bei dem die Genexpression in den Hyphenpilzen P. confluens und S. macrospora verglichen wird. P. confluens gehört zu einer ursprünglichen Gruppe von Ascomyceten und bildet einen eher einfachen Fruchtkörper, der als Apothezium bezeichnet wird, wohingegen S. macrospora zu einer abgeleiteten Ascomycetengruppe gehört und einen komplexeren Fruchtkörpertyp (Perithezium) bildet. Ein Vergleich der Genexpression zwischen diesen beiden Arten kann daher dazu beitragen, Gene zu identifizieren, deren Expressionsmuster im Verlauf der Fruchtkörperevolution erhalten geblieben sind.
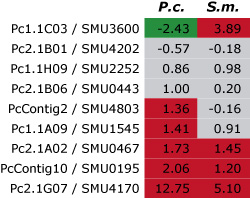
Dieses Bild zeigt das Ergebnis eines ersten Vergleichs der Genexpression für neun Gene (gekennzeichnet durch die jeweilige Nummer in jeder Zeile) von P. confluens (P.c.) und S. macrospora (S.m.). Für jedes Gen wurde das Verhältnis der Expression während der Fruchtkörperdifferenzierung im Vergleich zum vegetativen Wachstum mit Hilfe der quantitativen Real-Time-PCR ermittelt. Die Ergebnisse sind als logarithmische Werte angegeben, d.h. positive Werte (rot) zeigen an, dass ein Gen während der Entwicklung heraufreguliert ist, negative Werte (grün), dass es herunterreguliert ist. Werte zwischen -1 und +1 (grau) zeigen an, dass das Gen nicht differentiell exprimiert ist. Von den neun Genen sind drei in beiden Arten heraufreguliert und drei in beiden Arten nicht differentiell exprimiert. Dies zeigt an, dass die Genexpression während der Fruchtkörperentwicklung sogar in diesen beiden nicht sehr nah verwandten Pilzen zu einem hohen Grad konserviert ist.
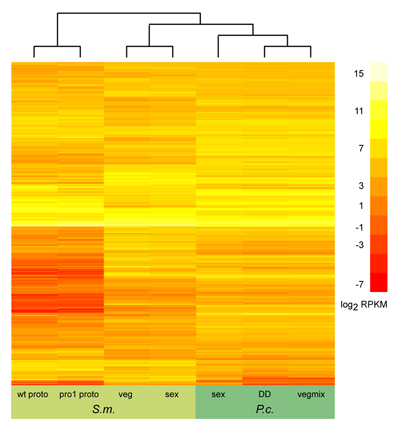
Links ist das Ergebnis eines ähnlichen
Vergleichs gezeigt, diesmal wurden allerdings alle orthologen Gene untersucht, die in den beiden Organismen unter verschiedenen
Bedingungen exprimiert werden (mehrere tausend Gene). Auf Grund der großen Zahl der untersuchten Gene sind die Gene nicht mehr
einzeln gekennzeichnet. Weiterhin zeigt dieser Vergleich keine Expressionsverhältnisse wie die Abbildung oben, sondern die
Zahl der Sequenz-Reads aus RNA-seq-Experimenten. Dieser Vergleich wurde im Rahmen des Pyronema-Genom- und Transkriptom-Projekts
durchgeführt, das weiter oben beschrieben wurde. Mehr Details gibt es in der entsprechenden Publikation.
- Abgesehen von evolutionär konservierten
Genexpressionmustern ist das sogenannten "Clustering", d.h. die Anordnung von Genen direkt nebeneinander im Genom und ihre gemeinsame Regulation, ein weiteres
wichtiges Kriterium, mit dem man bei Pilzen Gene identifizieren kann, die möglicherweise funktionelle Bedeutung haben. In Pilzen kodieren
solche Gene oft für Enzyme des sogenannten Sekundärstoffwechsels, bei dem verschiedenste Substanzen produziert werden, die oft
pharmakologisch relevante Wirkung zeigen. Zu den Sekundärmetaboliten gehören u.a. das Antibiotikum Penicillin, aber auch Gifte wie
das Mykotoxin Aflatoxin. Genomprojekte haben gezeigt, dass Pilze meist eine große Anzahl von Genclustern für Sekundärmetaboliten
enthalten, aber die Funktion der möglicherweise produzierten Stoffe ist größtenteils noch völlig unbekannt. Eine Möglichkeit ist,
dass Sekundärmetabolite unter bestimmten Bedingungen oder während bestimmter Lebensstadien für den Pilz nützlich sind, so z.B. während
der sexuellen Entwicklung. Daher haben wir eine "comparative functional genomics"-Analyse durchgeführt, bei der wir speziell nach
Genen gesucht haben, die nicht nur während der sexuellen Entwicklung in S. macrospora und N. crassa hochreguliert sind, sondern die
weiterhin auch physikalisch geclustert in den jeweiligen Genomen vorliegen.
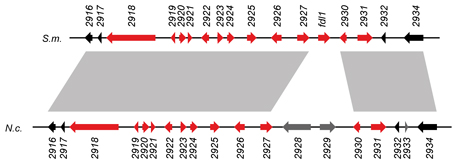
Das Bild zeigt einen solchen Cluster mit
der Genomorganisation in S. macrospora (S.m.) und N. crassa (N.c.). Die Gene sind durch Pfeile repräsentiert, die Farbe
zeigt an, ob das Gen während der sexuellen Entwicklung hochreguliert ist (rot) oder nicht (schwarz oder grau). Genregionen, die
durch graue Schattierung zwischen den beiden Spezies markiert sind, sind ähnlich. Dies zeigt, dass nicht nur die Genexpressionsmuster,
sondern auch die Anordnung der Gene im Genom zum größten Teil konserviert ist. Es konnte in einer genaueren Analyse gezeigt werden, dass eines der Gene (2925)
an der Fruchtkörperbildung beteiligt ist, da die Deletion dieses Gen zu verzögerter Fruchtkörperreifung führt.
- In den vorigen Projekten wurde die Genexpression auf Transkriptebene mit Hilfe von Mikroarrays und Real-Time-PCR untersucht. Genexpression kann allerdings noch auf vielen weiteren Ebenen reguliert werden, und auch diese können während der pilzlichen Entwicklung konserviert sein, wie die Analyse eines Fruchtkörper-spezifischen Proteins aus S. macrospora und N. crassa zeigte. Das Protein wurde entdeckt, weil es in diesen beiden Ascomyceten sehr stark in Fruchtkörpern angereichert wird, daher wurde es APP für "abundant perithecial protein" genannt.
-
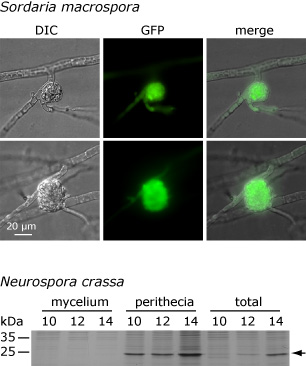
Dieses Bild zeigt, dass das APP-Protein sowohl in S. macrospora als auch in N. crassa nur in Fruchtkörpern und nicht im umgebenden Myzel angereichert wird. Der obere Teil zeigt die mikroskopische Analyse eines Fusionsproteins, das aus APP und dem grün-fluoreszierenden Protein (GFP) besteht. Das Fusionskonstrukt wurde unter der Kontrolle der regulatorischen Regionen von app exprimiert. Das linke Bild zeigt ein einen jungen Fruchtkörper (Protoperithezium) mit umgebenden Hyphen im differentiellen Interferenzkontrast (DIC). Die grüne GFP-Fluoreszenz kann im jungen Fruchtkörper beobachtet werden, nicht aber in den umgebenden Hyphen. Im unteren Teil ist die Analyse des nativen APP aus N. crassa zu sehen. Proteinextrakte aus Fruchtkörpern und dem umgebenden Myzel (total) sowie aus Fruchtkörpern allein (perithecia) bzw. dem umgebenden Myzel allein (mycelium) wurden nach 10, 12 oder 14 Tagen extrahiert und auf einem denaturierenden Proteingel aufgetrennt. Die APP-Bande ist durch einen Pfeil markiert. APP ist im Gesamtextrakt (total) und in den Fruchtkörpern zu sehen, aber nicht im umgebenden Myzel. Diese und andere Analysen zeigen, dass die Expression von app in S. macrospora und N. crassa sowohl zeitlich als auch räumlich konserviert ist. Eines unserer weiteren Ziele ist es herauszufinden, in welchem Grad die Genexpression während der pilzlichen Entwicklung auf verschiedenen regulatorischen Ebenen konserviert ist.
3. Analyse der Evolution der sexuellen Entwicklung in Tremellomyceten
- Tremellomyceten gehören zu den Basidiomyceten, zu denen unter anderem Speisepilze wie Champignons gehören. Allerdings sind die Tremellomyceten eine evolutionär früh entstandene Gruppe, in der nur wenige Arten vielzellige Strukturen bilden, die meisten wachsen als Einzeller oder wenigzellige Myzelien. Zu den Tremellomyceten gehören einige humanpathogene Pilze wie z.B. Cryptococcus neoformans, aber auch viele saprotrophe Pilze. Ein wichtiger Aspekt der Genomevolution der Tremellomyceten ist die Evolution der sexuellen Entwicklung, hier werden die Tremellomyceten als Modellorganismen untersucht. Vergleichende Analysen haben einen Trend hin zu großen Kreuzungstyp-Regionen im Genom gezeigt, ähnlich der Evolution von Sex-Chromosomen in Tieren und Pflanzen (Heitman et al. 2013, Mycologia 105: 1-27). Hierbei fusionieren bei Basidiomyceten der HD-Locus und der P/R-Locus zu einem großen Kreuzungstyp-Locus. Es ist allerdings noch unklar, wie weit verbreitet dieser Trend innerhalb der Tremellomyceten ist. Daher haben wir in einer Kooperation mit den Forschergruppen von Robert Kourist (Ruhr-Universität Bochum), Thomas Brück (TU München), Joseph Heitman (Duke University) und dem Joint Genome Institute das Genom des Tremellomyceten Trichosporon oleaginosus sequenziert, der ein Mitglied einer noch wenig untersuchten Tremellomyceten-Gruppe ist. In einer anschließenden Studie haben wir weitere Tremellomyceten-Genome analysiert, um festzustellen, wie weit verbreitet der evolutionäre Trend zu größeren Kreuzungstyp-Regionen ist. Dabei zeigte sich, dass alle untersuchten Genome aus der Ordnung Trichosporonales fusionierte Kreuzungstyp-Loci besitzen.
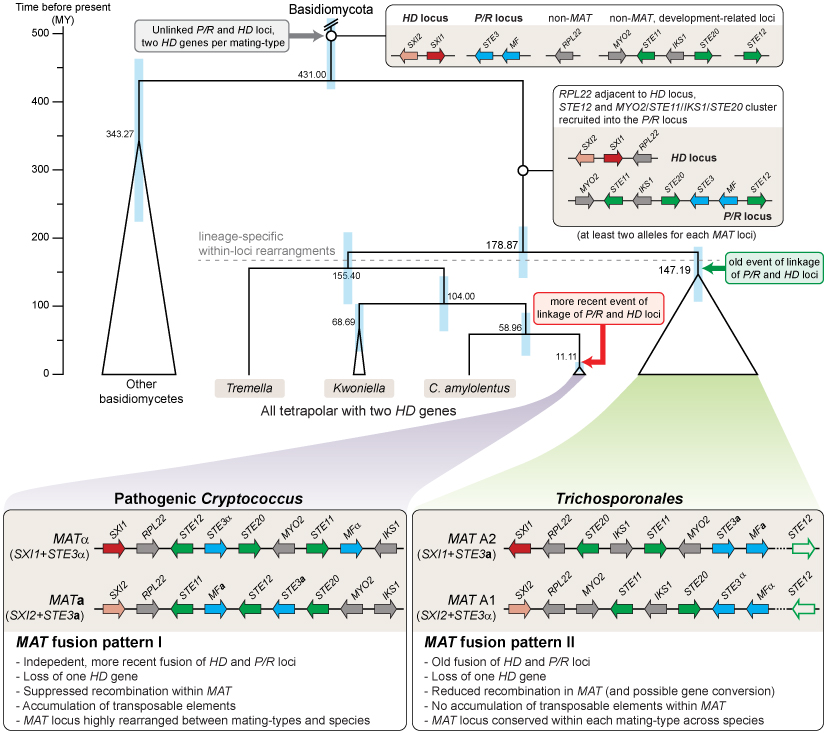
Die Abbildung zeigt ein Modell für die Evolution der Kreuzungstyp-Loci in Tremellomyceten, das auf Basis der vergleichenden Analyse der Genome von 24 Trichosporonales-Arten entwickelt wurde. Gene in den Kreuzungstyploci, die zu den Homöodomänen-Transkriptionsfaktoren (HD-Locus) oder den Pheromon- oder Rezeptorgenen (P/R-Locus) gehören, sind in rot/rosa bzw. blau dargestellt. Gene, die an der sexuellen Entwicklung beteiligt sind, aber nicht ursprünglich Teil des MAT-Locus waren, sind in grün dargestellt, andere Gene in grau. Es werden nur Gene gezeigt, die in C. neoformans und in den Trichosporonales mit den MAT-Genen (STE3, HD) gekoppelt sind (STE11, STE12, STE20, IKS1, MYO2, RPL22), andere Gene sind der Übersichtlichkeit halber nicht dargestellt. In pathogenen Cryptococcus-Arten und in den Trichosporonales kam es unabhängig zu einer Fusion der beiden Kreuzungstyp-Loci. Ebenso geschah der Verlust eines HD-Gens geschah zweimal unabhängig voneinander in Cryptococcus und den Trichosporonales. Weitere Details sind in der entsprechenden Veröffentlichung in PLoS Genetics zu finden (Sun et al. 2019, PLoS Genet 15: e1008365).